RESEARCH
Current and proposed research plan
Despite extensive research, psychiatric disorders remain poorly understood, mainly because their pathophysiology is far too complex to be elucidated using previous technology. Recent works have identified contributory factors for psychiatric disorders to be susceptible gene variants (molecular layer), synaptopathy (subcellular and cell layer), and alteration in neuronal circuits (circuit layer), which probably results in behavioral manifestations (individual layer). However, the relationship between each layer remains unknown, which hinders the integrative and causal mechanistic understanding of behavior. It seems likely that each layer can affect one another, from the macroscale to the mesoscale and then to the microscale layer, or vice versa. Thus, my team aims to gain a constructive understanding of the multiscale hierarchical nature of psychiatric disorders.
Project#1: Optical interrogation of synaptic effects on the upper layers of the brain
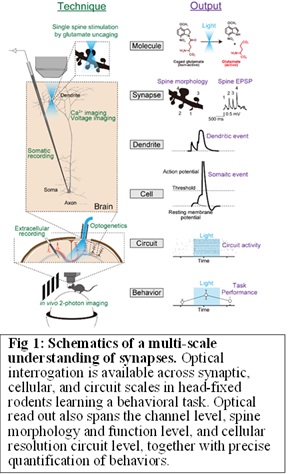 Our previous work demonstrated that a DISC1 knockdown (KD) mouse model of schizophrenia exhibited a decrease in dendritic spine density (Hayashi-Takagi et al., 2010, Nat Neurosci ; Hayashi-Takagi et al., 2014, Proc Natl Acad Sci USA ). We also found a significantly higher number of extra-large (XL) spines (> 0.7 μm in fixed samples) in this model compared to wild-type mice. The presence of XL spines mirrors findings from other established schizophrenia models, Calcineurin KO mice (Zeng H et al, 2001, Cell ) and SETD1A hKO mice (Nagahama et al, 2019, Cell Rep ). Thus, we aimed to elucidate the relationship among XL spines, neuronal computation, and working memory performance of SETD1A hKO and DISC1 KD mice in a multiscale manner (Fig 1). We found that XL synapses, which evoked supralinear dendritic and somatic integration, resulting in increased neuronal firing. The probability of XL spines correlated negatively with working memory, and the optical prevention of XL spine generation restored working memory impairment. The currently dominant hypothesis for schizophrenia pathophysiology is hypo-connection due to reduced synapse density. However, our results suggest that certain subsets of psychiatric disorders are also shaped by extra-strong synapses, which warrants the revision of schizophrenia pathophysiology for mechanism-oriented therapeutic strategies (Obi-Nagata et al, Science Advances , 2023). Now, we are investigating the relationship between synaptic activity (axonal Ca2+ imaging, iGluSnFR), neuronal firing (somatic Ca2+ imaging), and behavior by combining in vivo 2-photon imaging in a mouse model of disease (Setd1A hKO, Chd8 hKO, and MeCP2 hKO).
Our previous work demonstrated that a DISC1 knockdown (KD) mouse model of schizophrenia exhibited a decrease in dendritic spine density (Hayashi-Takagi et al., 2010, Nat Neurosci ; Hayashi-Takagi et al., 2014, Proc Natl Acad Sci USA ). We also found a significantly higher number of extra-large (XL) spines (> 0.7 μm in fixed samples) in this model compared to wild-type mice. The presence of XL spines mirrors findings from other established schizophrenia models, Calcineurin KO mice (Zeng H et al, 2001, Cell ) and SETD1A hKO mice (Nagahama et al, 2019, Cell Rep ). Thus, we aimed to elucidate the relationship among XL spines, neuronal computation, and working memory performance of SETD1A hKO and DISC1 KD mice in a multiscale manner (Fig 1). We found that XL synapses, which evoked supralinear dendritic and somatic integration, resulting in increased neuronal firing. The probability of XL spines correlated negatively with working memory, and the optical prevention of XL spine generation restored working memory impairment. The currently dominant hypothesis for schizophrenia pathophysiology is hypo-connection due to reduced synapse density. However, our results suggest that certain subsets of psychiatric disorders are also shaped by extra-strong synapses, which warrants the revision of schizophrenia pathophysiology for mechanism-oriented therapeutic strategies (Obi-Nagata et al, Science Advances , 2023). Now, we are investigating the relationship between synaptic activity (axonal Ca2+ imaging, iGluSnFR), neuronal firing (somatic Ca2+ imaging), and behavior by combining in vivo 2-photon imaging in a mouse model of disease (Setd1A hKO, Chd8 hKO, and MeCP2 hKO).
Project #2: Establishment of a novel synaptic imaging for functional connectomics
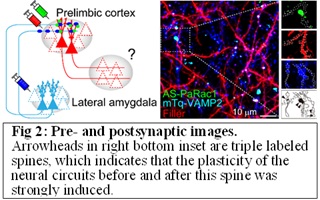 We previously developed a synaptic optoprobe, Activated Synapse targeting PhotoActivatable Rac1 (AS-PaRac1). AS-PaRac1 is unique; not only can it specifically label recently potentiated dendritic spines, but it can also selectively induce shrinkage in spines containing AS-PaRac1 (Hayashi-Takagi et al, 2015, Nature ). However, the biggest drawback of AS-PaRac1 is that it only labels potentiated spines, which doesn't provide the circuit information. To overcome this issue, we established a novel functional connectomics imaging: a tri-colour imaging method of activity-dependent expression of the presynaptic marker Vamp2, AS-PaRac1, and posynaptic neurons in vivo brain (Fig 2). Now, a visualization of the 'synaptic and circuit ensemble' in the psychiatric condition is underway. Together with synaptic shrinkage by AS-PaRac1 experiments (a projection pathway-specific optogenetics), we now try to elucidate the dynamic nature of synaptic potentiation in a specific neuronal circuit chosen from the vastly complex brain (functional connectomics). We are also working on establishing a new method of photo-sensitive circuit tracing, which we cannot explain here yet. If you are interested, please contact Hayashi directly. In addition, a combination of these methods with the Project #3 (refer to Project #3 below) and TissueCyte in CBS core facility, we aim to perform a comprehensive analysis of how such potentiation is distributed throughout the brain in control and disease mouse models at a single-synapse resolution.
We previously developed a synaptic optoprobe, Activated Synapse targeting PhotoActivatable Rac1 (AS-PaRac1). AS-PaRac1 is unique; not only can it specifically label recently potentiated dendritic spines, but it can also selectively induce shrinkage in spines containing AS-PaRac1 (Hayashi-Takagi et al, 2015, Nature ). However, the biggest drawback of AS-PaRac1 is that it only labels potentiated spines, which doesn't provide the circuit information. To overcome this issue, we established a novel functional connectomics imaging: a tri-colour imaging method of activity-dependent expression of the presynaptic marker Vamp2, AS-PaRac1, and posynaptic neurons in vivo brain (Fig 2). Now, a visualization of the 'synaptic and circuit ensemble' in the psychiatric condition is underway. Together with synaptic shrinkage by AS-PaRac1 experiments (a projection pathway-specific optogenetics), we now try to elucidate the dynamic nature of synaptic potentiation in a specific neuronal circuit chosen from the vastly complex brain (functional connectomics). We are also working on establishing a new method of photo-sensitive circuit tracing, which we cannot explain here yet. If you are interested, please contact Hayashi directly. In addition, a combination of these methods with the Project #3 (refer to Project #3 below) and TissueCyte in CBS core facility, we aim to perform a comprehensive analysis of how such potentiation is distributed throughout the brain in control and disease mouse models at a single-synapse resolution.
Project #3: Development of a novel dual-axis 2-photon imaging with synaptic resolution
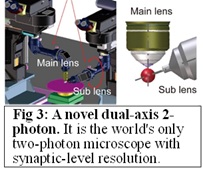 To further accelerate the results of Project #1 and 2, we created a high-resolution (synaptic resolution) two-axis two-photon microscopy system capable of imaging two brain regions simultaneously (Fig 3). Together with two-colour calcium imaging, using a different coloured in each area, we will determine how information processing, such as feed-forward or coupling, is altered in the psychiatric model mice.
To further accelerate the results of Project #1 and 2, we created a high-resolution (synaptic resolution) two-axis two-photon microscopy system capable of imaging two brain regions simultaneously (Fig 3). Together with two-colour calcium imaging, using a different coloured in each area, we will determine how information processing, such as feed-forward or coupling, is altered in the psychiatric model mice.
Project #4: Synaptic assay in the human specimens
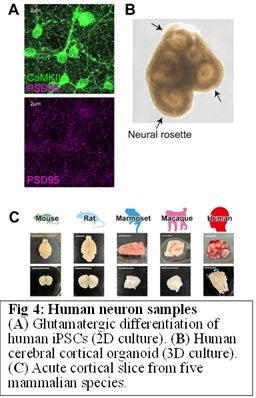 The next question is whether this phenomenon can be detected in the brains of human psychiatric patients and whether it is indeed the cause of the disorders. To bridge the gap between mouse findings to human psychiatric disorders, we started to use postmortem brain from psychiatry patients (From Pittsburg Univ and Japan Brain Bank Network), human iPS cell-derived neurons with disease-related gene mutations (Fig 4AB ), and neurosurgically removed samples (Fig 4C, RIKEN CBS-Keio Univ collaboration). For a detailed plan, please refer to “10-year research vision”.
The next question is whether this phenomenon can be detected in the brains of human psychiatric patients and whether it is indeed the cause of the disorders. To bridge the gap between mouse findings to human psychiatric disorders, we started to use postmortem brain from psychiatry patients (From Pittsburg Univ and Japan Brain Bank Network), human iPS cell-derived neurons with disease-related gene mutations (Fig 4AB ), and neurosurgically removed samples (Fig 4C, RIKEN CBS-Keio Univ collaboration). For a detailed plan, please refer to “10-year research vision”.
10-year research vision
Our research has focused on mouse models of psychiatric disorders, and we plan to continue this strategy. However, this strategy alone will not achieve the elucidation of human psychiatric disorders. The pathophysiology of psychiatric disorders could be answered, for example, by using ,in vivo electrophysiological techniques to measure neuronal firing in schizophrenic patients and correlate it with auditory hallucinations, and then, having identified the synapses that might be involved, to test whether optical manipulation of these synapses can manipulate auditory hallucinations. This is ethically impossible. However, it should be possible to overcome the limitations of each method alone, to deepen our understanding of the pathophysiology of mental disorders. I believe that the combinatorial use of human-derived neurons, theoretical neuroscience, and reverse translational feedback to the mouse research is key. Human-derived samples include postmortem brains, human iPSCs-derived neurons, and neurosurgically removed acute brain samples. Although functional experiments are not possible in postmortem brains, it is possible to morphologically study the synaptic properties of the psychiatric group and age-matched controls. iPS-derived neurons are not perfect neurons, but it is easy to perform genetic manipulation on these cells. For example, introducing disease-related genetic mutations and rescue experiments are easy, and we can test many hypotheses with a sufficient number of experiments. In addition, to measure the ground truth of the human neurophysiological properties, we use glioma samples obtained for therapeutic purposes at the Department of Neurosurgery at Keio University. Since gliomas are highly invasive and it is therapeutically essential to remove the surrounding healthy neural tissue, healthy neurons can be ethically obtained. Indeed, preliminary experiments with these samples have shown that whole-cell patch clamping and 2-photon experiments at the single synapse level are possible. Finally, although such human data can provide invaluable insights, they do not by themselves lead to an understanding of human disorders. Therefore, to bridge the gap between the obtained "human data" and "our questions", we will use computational methods. We will integrate the human data to mathematical model, specifically the NEURON model, the Leaky Integrate-and-Fire model, and the Free Energy model, and construct neural, circuit, and behavioral models that implement the human parameters. These simulations will provide new hypotheses for future experiments in mouse studies. I believe that a rapid circulation between mouse, human, and theoretical neuroscience is crucial to understanding human psychiatric disorders, and my lab will conduct research based on this strategy for the next 10 years.
MEMBER
HAYASHI-TAKAGI Akiko

- Team Director
- akiko.hayashi-takagi[at]riken.jp
- Former Psychiatrist
- Project director of MEXT Grant-in-Aid for Scientific Research on Innovative Area “Multi-scale brain (FY2018-2022)” Dedicate all my passion for the elucidation of psychiatric disorders
TSUTSUMI Shinichiro

- Deputy Team Director
- shinichiro.tsutsumi[at]riken.jp
KOH Isabel Siew Yin

- Research Scientist
- isabelsiewyin.koh[at]a.riken.jp
OBI-NAGATA Kisho

- Visiting researcher
- Assistant professor at Gunma Uni.
- kisho.obi[at]a.riken.jp
OKIMURA Tsukasa

- Visiting researcher
- Lecturer at Showa Uni.
- tsukasa.okimura[at]a.riken.jp
Suzuki Norimitsu

- Technical Staff I
- norimitsu.suzuki[at]riken.jp
OZAWA Katsuya

- Technical Staff I
- katsuya.ozawa[at]riken.jp
HISANO Yasuko

- Technical Staff I
- yasuko.hisano[at]riken.jp
SATO Miki

- Technical Staff I
- miki.satoh[at]riken.jp
EBISUI Etsuko

- Technical Staff II
- Etsuko.ebisui[at]riken.jp
ISODA Kiichiro

- Technical Staff
- kiichiro.isoda[at]riken.jp
Our Address
Multi-scale Biological Psychiatry
RIKEN Center for Brain Science
- 2-1 Hirosawa, Wako, Saitama 351-0198 Japan.
- +81-48-462-1111
- hayashi_lab[at] ml.riken.jp (Replace [at] with @ )
- multi-scale_psychiatry.riken.jp